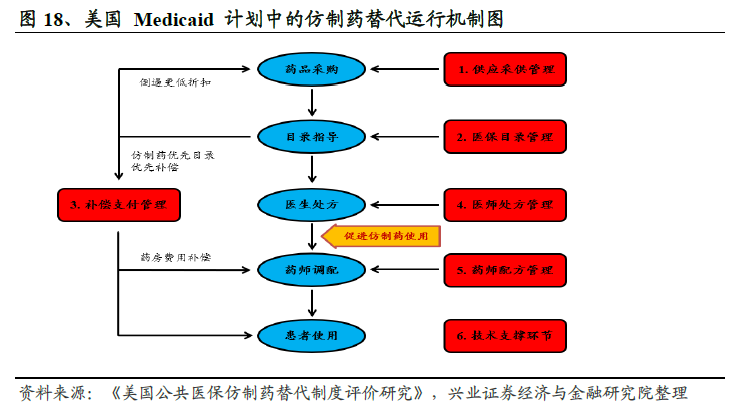
美國仿制替代原研之路。近年來,全球藥品市場迅速發展,根據前瞻產業研究院《中國醫藥行業“十三五”市場前瞻與發展規劃分析報告》的數據統計,2016 年全球醫藥市場規模達到11100億美元,2011-2016 年復合增長率高達6%,美國市場占比40%以上。根據IMS 數據庫統計,2016 年全球仿制藥市場規模達到2200 億美元,美國是全球最大的仿制藥消費國,2016 年美國市場仿制藥銷售總額約為800 億美元。Evaluate Pharma預計未來幾年,美國仿制藥市場年復合增長率將達到9.1%,2020 年仿制藥銷售額將突破1100 億美元。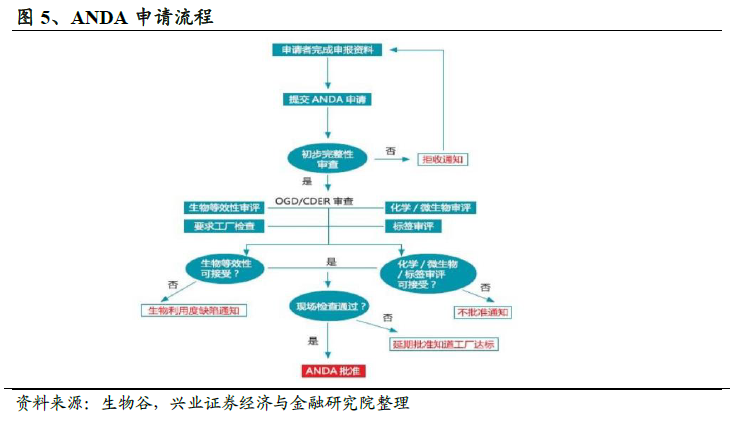
由于全球人口結構老齡化的加深、新藥價格越趨昂貴,使得醫保支付壓力日益沉重,多國政府開始積極鼓勵使用仿制藥。美國的醫療衛生費用支出位居全球第一,2016 年美國醫療衛生總支出達3.3 萬億美元,占GDP 的17.9%。低價仿制藥的普及給美國醫療衛生體系帶來了可觀的開支節約,據統計,過去十年共節省了16700億美元,其中2016 年節省2530 億美元,超過當年仿制藥銷售額。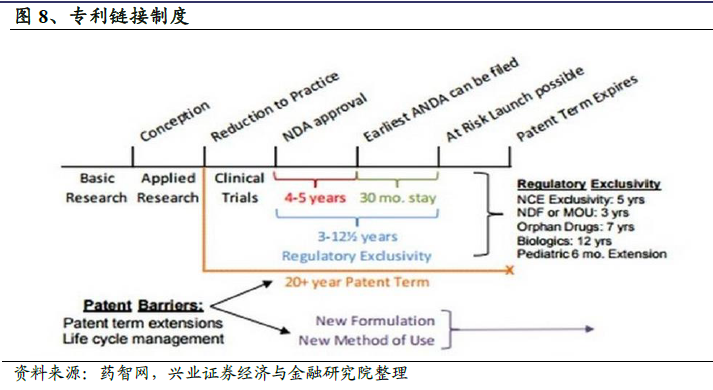
寬松管理,初步萌芽。上世紀初,美國藥品上市要求與一般商品相同,無需安全性與有效性的證明。1938年,美國一家公司生產的抗炎癥藥物造成醫療事故,此后美國政府要求上市藥品必須具備安全性并向FDA 申報,但只需提交公開文獻資料證明安全性。直到二戰之前,美國制藥市場基本由歐洲生產商壟斷,二戰期間為了滿足戰爭用藥要求,美國仿制藥行業逐漸興起,并在戰爭結束后繼續依靠歐洲原研藥賺取利潤。
嚴格管控,緩慢推進。1962 年以前,FDA 只負責審批藥品的安全性。1961 年,在歐洲、加拿大等多個國家上市的“反應停”藥物被指出造成上萬名嬰兒畸形,該事件促使美國政府于1962 年通過《Kefauver-Harris 修正案》,從此之后,新藥在上市前,必須要向FDA提供證明其安全性及有效性的數據,修正案規定仿制藥須在原研藥的專利全部到期后才可以進行研制,同時所有新藥申請不僅需要證明其安全性,還需要提供證明有效性的臨床研究數據,由此帶來的上市時間延長與臨床研究高額費用導致仿制藥市場發展緩慢,極大的限制了研制仿制藥的商業動力。至1983 年,專利到期的原研藥中只有35%存在相應的仿制藥競爭,且市場份額平均僅有13%。與此同時,藥物價格高漲,給政府、保險公司、民眾帶來了巨大的經濟負擔。
有序監管,高速發展。在此背景下,《Hatch-Waxman 法案》(《藥品價格競爭和專利期保護法》,俗稱《仿制藥法》)于1984 年9 月由美國國會簽署通過,自1984 年11 月起正式實施,該法案創造了仿制藥的現代審批體系。對于仿制藥而言,該法案采用了簡化新藥申請(Abbreviated New Drug Application,簡稱ANDA),以生物等效性(Bioequivalence,BE)試驗代替此前的臨床試驗,Hatch-Waxman 法案假設生物等效性是藥品安全性和有效性的一個良好的替代指標;并允許仿制藥在原研藥專利到期前開始研發,大幅降低了仿制藥研發上市所需的時間與費用。